Title: The Cloud and the Shell - Applied Bioinformatics on the Example of Gene Expression Analysis using Unix and freely available Open Source Tools
Author: Alexander Kerner
EMail: training at silico-sciences.com
Seminar Ruprecht-Karls-Universität Heidelberg
[TOC]
nohup: run a command immune to hangups, with output to a non-tty
$ nohup p1 &
[1] 14958
$ less nohup.out
-
Get and save the key file
-
cdto the corresponding directory -
'Install' the ssh key
$ ssh-add training@silico_rsa Enter passphrase for training@silico_rsa: Identity added: training@silico_rsa (training@silico_rsa) -
In case of permissions error, fix permissions
$ chmod 600 training@silico_rsa -
In case of an ssh-agent error, fix as described here
ssh: OpenSSH SSH client (remote login program)
$ ssh silico-sciences.com
$ ssh 176.28.21.178
$ ssh [yourname]@silico-sciences.com
-
Login to the remote system using
[yourname]@silico-sciences.com -
Verify successful login:
-
whoami -
hostname -
ifconfig -
Get some system infos:
$ cat /etc/lsb-release DISTRIB_ID=Ubuntu DISTRIB_RELEASE=14.04 DISTRIB_CODENAME=trusty DISTRIB_DESCRIPTION="Ubuntu 14.04.3 LTS" -
Type these commands on the remote system and on the local system.
-
Use wget to download data from here.
There are many ways to retrieve all files in that list, here are some hints:
-
Download this list as a file.
-
Use
sedortrto replace all new line characters (\n) with a space character ( -
Pipe the resulting list to a file or directly to
wget. -
Take a look at
wgets-ioption. -
Use
wgetwith the--no-directories, the--accept-regexand the--recursiveoption.
TopHat uses Bowtie2 as a 'mapping engine'. Bowtie2 requires the reference genome to be indexed.
Create this index file as described here.
-
Use
Tophatto map the reads to the reference genome:$ tophat -o [some-out-dir] -G [reference-annotation].gtf [reference-bowtie2-index-file] [reads]_1_fastq [reads]_2_fastq `Tophat` produces several output files: 0. `accepted_hits.bam`Note: See here howto avoid repetitive index building. Find a pre-build transcriptome index for chromosome three here
/var/data/bi/reference/prebuild/Homo_sapiens/Ensembl/GRCh37/Annotation/Genes/transciptome_index/genes_chr03. -
Use
samtools idxstatsto see the number of mapped/ unmapped reads in the createdaccepted_hits.bamfile (see here).$ samtools idxstats accepted_hits_sorted.bam | column -t 3 198022430 14926 0 * 0 0 0 $ samtools idxstats unmapped_sorted.bam | column -t 3 198022430 0 0 * 0 0 2 -
Take a look at the
bamfiles:-
less accepted_hits.bam -
zless accepted_hits.bam -
samtools view accepted_hits.bam
-
-
Use
Cuffquantto precompute gene expression levels.$ cuffquant [reference-annotation].gtf [tophat_out]/accepted_hits.bamOptions (less speed, more accuracy):
-
-b/--frag-bias-correct: use bias correction - reference fasta required -
-u/--multi-read-correct: use 'rescue method' for multi-reads$ cuffquant -b [reference-seq].fa -u [reference-annotation].gtf [tophat_out]/accepted_hits.bam
-
-
Use
Cuffdiffto find significant changes in expression level.$ cuffdiff -o [some-out-dir] -L Lung,Stomach,Heart [reference-annotation].gtf [lung1-4-cuffquant_out]/abundances.cxb,[lung2-4-cuffquant_out]/abundances.cxb,[lung3-4-cuffquant_out]/abundances.cxb,[lung4-4-cuffquant_out]/abundances.cxb [stomach1-4-cuffquant_out]/abundances.cxb,[stomach2-4-cuffquant_out]/abundances.cxb,[stomach3-4-cuffquant_out]/abundances.cxb,[stomach4-4-cuffquant_out]/abundances.cxb [heart1-4-cuffquant_out]/abundances.cxb,[heart2-4-cuffquant_out]/abundances.cxb,[heart3-4-cuffquant_out]/abundances.cxb,[heart4-4-cuffquant_out]/abundances.cxb $ cuffdiff -o [some-out-dir] -L Lung,Heart [reference-annotation].gtf [lung1-4-cuffquant_out]/abundances.cxb,[lung2-4-cuffquant_out]/abundances.cxb,[lung3-4-cuffquant_out]/abundances.cxb,[lung4-4-cuffquant_out]/abundances.cxb [heart1-4-cuffquant_out]/abundances.cxb,[heart2-4-cuffquant_out]/abundances.cxb,[heart3-4-cuffquant_out]/abundances.cxb,[heart4-4-cuffquant_out]/abundances.cxbNote: Pay attention to correct usage of commas and spaces. Separate replicates with commas (don't use
,[whitespace]) and conditions/ labels with space.
Cuffdiff writes fold changes to the table [cuffdiff_out]/genes_exp.diff.
-
Use
cutto cut away columns that we are not interested in. -
Use
sortto sort the table by-
significance and
-
absolute log2 fold change (descending).
-
-
use
grepand|to extract lines with a significant regulation into a new file.
Do not override [cuffdiff_out]/genes_exp.diff. Use pipes instead!
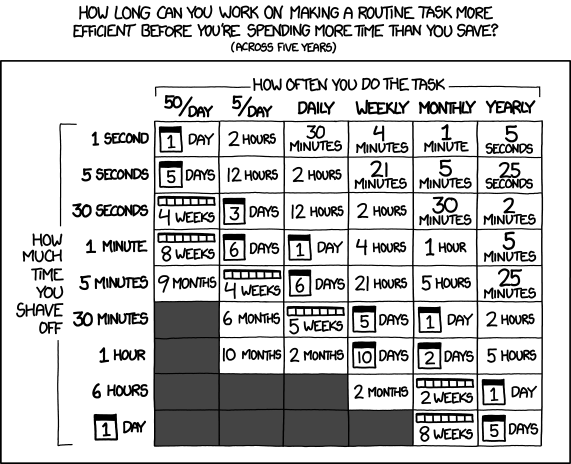
If you ask yourself if it is worth to write a script or not, take a look at this matrix: